近日,山东大学微生物技术国家重点实验室的史大永教授团队在药物化学领域的顶级期刊《Journal of Medicinal Chemistry》上以封面论文形式(封面设计灵感源于中国古老神话“后羿射日”)在线发表了题为“Design of selective PARP-1 inhibitors and antitumor studies”的研究论文。硕士研究生张依婷和李祥乾副研究员共同担任该论文的第一作者,史大永教授为唯一通讯作者,山东大学微生物技术国家重点实验室作为第一完成单位和通讯作者单位,为这项研究提供了坚实的支持。研究团队深入分析了PARP-1和PARP-2的氨基酸序列,发现了一个具有巨大潜力的选择性位点——S site。该位点由α-5螺旋和D-环上几个独特的氨基酸残基组成,为开发选择性PARP-1抑制剂提供了新的方向。基于这一S位点以及课题组长期对卤代化合物的研究,设计并合成了一系列活性化合物,并对其选择性以及抑制PARP-1酶活性最好的化合物I16开展了多维度的抗肿瘤活性评价与作用机制研究。

PARP(Poly (ADP-ribose) polymerase)在DNA损伤修复和基因组稳定性维护中占据核心地位。尽管PARP-1抑制剂的研究已历经半个世纪,市场上已有Olaparib、Niraparib、Rucaparib、Talazoparib、Fluzoparib和Pamiparib六种药物获准上市,但它们的临床应用却受限于血液学毒性。研究表明,这些副作用可能与PARP-1和PARP-2的同时抑制有关。实际上,仅针对PARP-1蛋白的抑制已足够展现抗肿瘤活性,且毒性更低。因此,开发高选择性的PARP-1抑制剂已成为当前研究的热点。当前大多数的PARP-1抑制剂设计都模仿NAD+的结构,通过与PARP-1的烟酰胺-核糖结合位点(NI位点)竞争性结合来发挥作用。然而,由于PARP-1和PARP-2的NI位点高度保守,仅针对NI位点的设计难以获得高选择性的抑制剂。为了突破这一瓶颈,科研团队需要寻找新的策略和设计方法,以开发出既安全又高效的选择性PARP-1抑制剂,为癌症治疗带来新的希望。
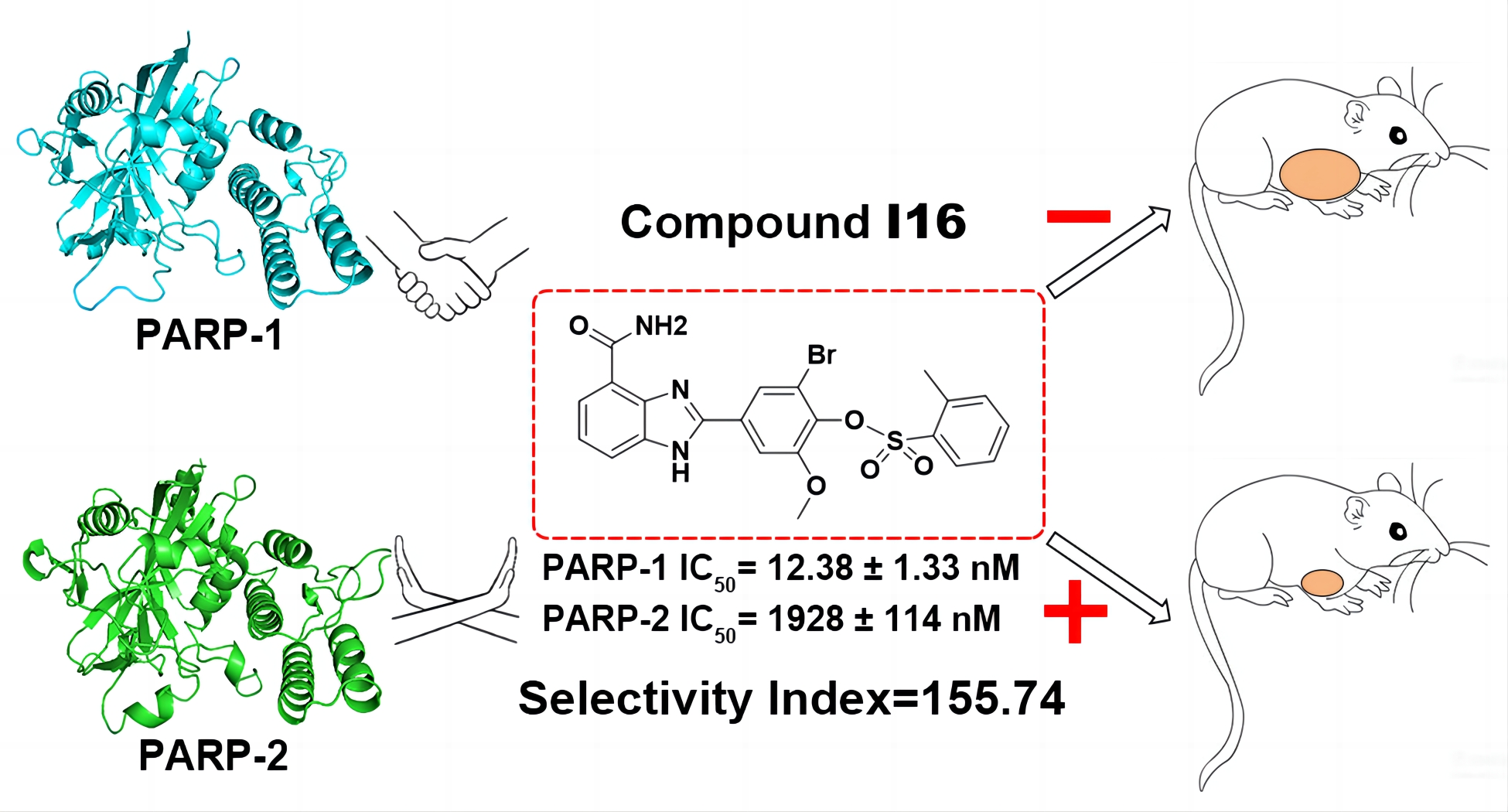
研究团队通过细致比较PARP-1与PARP-2催化结构域附近的氨基酸序列,成功发现了一个潜在的高选择性药物设计位点——S sie。这一位点由α-5螺旋和D-环上的几个独特氨基酸残基组成。基于这一发现,团队精心设计了溴苯酚-硫代氨基甲酮衍生物和溴酚-苯并咪唑-4-甲酰胺衍生物,并深入研究了它们对PARP-1/2的抑制活性。实验结果显示,与先导化合物B4相比,新型化合物I16在PARP-1酶抑制活性上提高了3倍,同时对PARP-2的选择性也显著增强,达到4倍(IC50=12.38±1.33 nM,选择性=155.74)。进一步的研究表明,I16能有效抑制SK-OV-3细胞的增殖、生长和迁移,并诱导其凋亡。在动物实验中,口服I16(25 mg/kg)对Hela和SK-OV-3肿瘤细胞异种移植模型的抑制率均显著高于口服阳性药物Olaparib(50 mg/kg)。更为引人注目的是,I16展现出了极高的安全性,即使在口服高剂量时也未观察到明显毒性。此外,研究团队还通过表面等离子共振(SPR)和酶活性抑制实验,验证了S位点中几个关键氨基酸的突变能够显著减少化合物与PARP-1蛋白的结合,进一步证实了S位点作为选择性PARP-1抑制剂设计新靶点的潜力。
该研究工作得到了国家重点研发计划、山东省自然科学基金、山东省重点研发计划、2023年青岛市新兴产业培育项目、山东省科技型中小企业创新能力提升工程项目以及中国科学院特别科研助理支持项目等科研项目的支持。山东大学微生物技术国家重点实验室生命环境研究公共技术平台为本工作提供了重要技术支持。
文章链接:https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c02460

